
新闻中心
推荐产品

 内容编辑
内容编辑嵌合抗原受体(Chimeric Antigen Receptor, CAR)设计的基础是T细胞活化的信号转导。T细胞受体(T-cell Receptor, TCR)检测由抗原递呈细胞(Antigen Presenting Cells)递呈的以主要组织相容性复合物(Major Histocompatibility Complex, MHC)-抗原肽复合体形式出现的抗原。T细胞受体与MHC-抗原肽复合体的结合诱导了胞内的级联反应:磷酸化的TCR募集胞内的第二信使提供第一个信号,T细胞表面的共刺激分子(CD28、CD27、CD134、CD137或ICOS)与抗原递呈细胞上各自相应的受体(CD80、CD86、CD137L或ICOSL)结合提供第二个信号。最终,T细胞被触发并活化,继而分泌穿孔素、颗粒酶和细胞因子,包括白介素2(IL-2)和干扰素γ(IFN-γ),通过诱导靶细胞凋亡防御感染。
然而,由于缺少MHC的表达以及肿瘤的弱免疫原性,正常的T细胞不能有效识别肿瘤。研究者在20世纪80年代中期第一次开发了嵌合免疫受体(CAR)。在1993年,Eshhar等在黑素瘤的治疗中对T细胞进行修饰以表达嵌合免疫受体(CAR),突破了MHC限制和弱免疫原性的问题。通常,嵌合免疫受体(CAR)由三个结构域组成:一个胞外的单链抗体片段(scFv),作为靶向部分通过与肿瘤相关抗原(Tumor-associated Antigens, TAAs)的特异结合使T细胞转向肿瘤细胞;一个跨膜区和一个胞内区,后者通常是由CD3ζ链和共刺激分子[如CD28和4-1BB(CD137)] 组成的信号转导结构域。根据胞内结构域的不同,CAR分为三代。第一代的胞内结构域仅包括一条CD3ζ链;第二代包括一条CD3ζ链和一个共刺激分子[CD28、4-1BB、CD134(OX40)或ICOS];第三代包括一条CD3ζ链和两个或更多个不同的共刺激分子。Zhang等比较了作为共刺激分子的CD28和4-1BB,并证明4-1BB对记忆性CD8+T细胞增加是必须的,并且在共刺激CD8+细胞毒淋巴细胞生成时优于CD28。因此,在嵌合免疫受体(CAR)设计时用4-1BB作为共刺激因子,有助于延缓钝化作用和增强CAR-T细胞的有效性。
鉴于CAR-T细胞治疗对非实体瘤前所未有的有效性,针对实体瘤的临床试验数量也在逐渐增加,尤其是抗-CD19 CAR-T细胞(表1)。CAR-T细胞治疗白血病取得成功的一个重要原因是,非实体肿瘤细胞在血液和淋巴系统中循环,更容易接触到获得性CAR-T细胞,并诱导其杀伤活性。然而在实体瘤中,由于存在多个障碍层(如细胞外基质)和缺少趋化因子,CAR-T细胞难于移动到肿瘤位点,而在实体瘤中常常与受体错配。由于抑制性微环境的存在(图1),即便有少部分CAR-T细胞成功抵达肿瘤位点,也有可能不再具有活性。另一个重要原因是,在实体瘤中很难发现特异的肿瘤相关抗原(TAA),如在B细胞急性淋巴母细胞性白血病中的CD19。因此, CAR-T细胞需要其他新型修饰以增强对实体瘤的有效性。
靶抗原的特异性
针对实体瘤的CAR-T细胞治疗的临床试验报告显示大部分CAR-T细胞治疗均面临“脱靶效应”(On-Target/Off-tumor)的困境。理想的肿瘤相关抗原需要在肿瘤细胞中特异表达;然而,一些肿瘤相关抗原也在正常细胞中表达。例如,间皮素不仅在间皮瘤中过表达,也在腹膜、胸膜和心包表面表达。另外,大多数肿瘤除去了其肿瘤相关抗原上的免疫原性抗原表位,以逃脱宿主免疫系统的攻击。因此,鉴别特异性免疫原性肿瘤抗原对于实体瘤的治疗是有必要的。
研究人员可以设计靶向异常修饰的肿瘤相关抗原或肿瘤特异性致瘤突变的嵌合抗原受体(CAR),如截短的MUC1。例如,Posey等最近报道了靶向异常糖基化的肿瘤相关的细胞膜粘蛋白(Cell Membrane Mucin, MUC1)新型嵌合抗原受体。在这项研究中,研究人员使用了以4-1BB作为共刺激分子、高亲和抗体(5E5)scFv区作为结合结构域的第二代嵌合抗原受体(CAR),该抗体靶向特异性出现在肿瘤组织中的截短O-糖肽抗原表位。因此,CAR-T细胞在正常情况下并不与糖基化的MUC1结合,但是在此例中它们特异识别肿瘤细胞上的MUC1的Tn糖型。该研究也证明MUC1-CAR-T细胞对正常的原代细胞不具有细胞毒作用。
表1 实体瘤CAR-T细胞治疗汇总
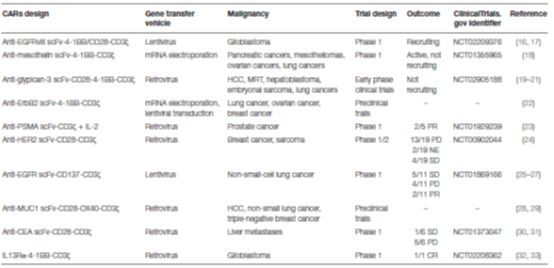
PD, progressive disease进行性疾病;PR, partial response部分响应; SD, stable disease 稳定的疾病;NE, not evaluable 无法评价;CR, complete remission;CAR, chimeric antigen receptor 嵌合抗原受体; scFv, single-chain antibody fragment 单链抗体片段;PSMA, prostate-specific membrane antigen前列腺特异性膜抗原;;IL-2, interleukin 2 白细胞介素2.。
研究人员也测试了特异表达于肿瘤细胞上的新抗原。新抗原是体细胞突变肿瘤细胞上产生的抗原,并且对每个患者的癌症都是独特的,此类抗原能够促进肿瘤生长和/或浸润。有研究人员建议利用新一代测序技术结合高通量的免疫学筛选方法鉴定免疫原性突变。研究者从患者肿瘤细胞中分离正常细胞,然后采用全组学和转录组学测序鉴定体细胞突变,这类突变可以被抗原递呈细胞递呈并激活免疫应答。Verdegaal等发现T细胞介导的新抗原免疫编辑和T细胞识别的新抗原的去表达可以引起肿瘤抗性。因此,在获得性T细胞治疗中监测新抗原的变化状况是必要的。
最近,一种新的嵌合抗原受体——串联嵌合抗原受体(Tandem CARs)——被设计出表达两个抗原结合结构域;串联CAR-T细胞仅在同时识别两种不同抗原时是被激活。改造的能同时靶向两种不同抗原的嵌合抗原受体更加特异和安全。例如,Hegde等将抗人表皮生长因子受体2(HER2)的scFv与结合IL-13能力突变的IL-13受体α2(IL-13Rα2)连接起来,并用CD28作为共刺激因子和CD3ζ链作为信号转导结构域,开发了一种新的串联嵌合抗原受体。该串联嵌合抗原受体具有结合HER2或IL-13Rα2的能力,防止肿瘤细胞侵染。与单一的CAR-T细胞相比,在HER2或IL-13α2出现时,这类CAR-T细胞的活化程度更容易维持但并不会耗尽。在小鼠胶质母细胞瘤模型中,串联CAR-T细胞减少抗原逃脱展示,增强抗肿瘤能力,并提高动物存活几率。
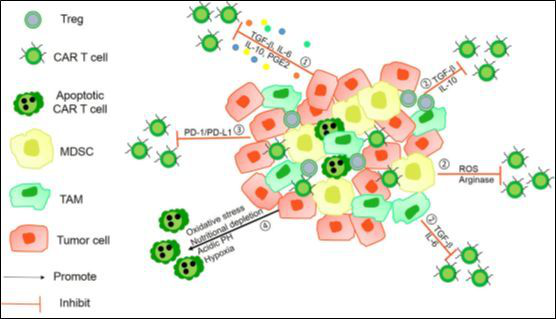
图1 肿瘤微环境的免疫抑制机制
①肿瘤来源的可溶性因子【如前列腺素E2(PEG2)】和细胞因子【如转化生长因子β(TGF-β)、IL-6和IL-10】抑制嵌合抗原受体(CAR)T细胞活性。
②免疫抑制性免疫细胞,即髓源性抑制细胞(MDSC)、调节性T细胞(Treg)、肿瘤相关的小噬细胞(TAM)或嗜中性粒细胞(TAN),通过Arg-I、ROS生成和一些可溶性抑制因子抑制T细胞功能。
③肿瘤细胞能够通过上调表面抑制受体,如程序性死亡配体1(PD-L1)/PD-L2,使用内在的负调控机制。
④不利的肿瘤微环境由于缺氧、氧化刺激、酸性pH和营养缺乏,使得CAR-T细胞难以存活。
另一种具有两种不同的scFv的串联CAR被设计出来,一个与CD3ζ链连接提供第一信号,另一个与共刺激分子连接提供第二信号。靶抗原单独表达不足以启动T细胞活化。只有两种抗原同时在靶细胞上表达才能激活CAR-T细胞,并诱导抗肿瘤效应。例如,研究人员提出一种即便无真正的肿瘤限制性抗原,仍能使CAR-T细胞对前列腺肿瘤具有特异性的方法。在此项研究中,研究者使用了两种前列腺肿瘤抗原——前列腺特异的膜抗原(Prostate-specific Membrane Antigen, PSMA)和前列腺干细胞抗原(Prostate Stem Cell Antigen, PSCA),并证明CAR-T细胞能破坏同时表达PSMA和PSCA的肿瘤细胞。在同源小鼠模型中,也显示CAR-T细胞被活化,并抵抗同时表达PSCA和PSMA的肿瘤。然而,在抗原特异T细胞的胁迫下,肿瘤细胞产生去表达抗原并具有抗性的新突变。研究者需要监测抗原的变化状况以改善CAR-T细胞治疗。
靶抗原的敏感性
敏感性是CAR-T细胞疗法治疗实体瘤的另一个挑战。T细胞和其靶细胞之间的空间距离在T细胞活化和信号转导中也扮演着关键角色。基于免疫受体络氨酸活化的模体被Src家族的淋巴细胞特异性激酶(Lck)磷酸化对于T细胞活化是必要的。然而,Lck在T细胞中起初是被抑制的,因而不具有磷酸化活性。它被蛋白络氨酸磷酸酶CD45和CD148激活,引起下游信号转导。一些研究表明,T细胞和抗原递呈细胞(APC)在形成免疫突触时之间的距离约为15 nm。早先的研究证明,在细胞界面去除CD45对于T细胞突触的形成是充分必要的。CAR-T细胞和其靶细胞间的空间距离可能也同等重要。然而,这依赖于完全不同的结构因素,包括scFv的空间结构、嵌合抗原受体在细胞膜上的位置以及抗原在靶细胞上的位置等。例如,Hombach等研究了特定抗原表位位置对CAR-T细胞活化的效率影响。他们证明,靶向近端表位而不是远端表位时,T细胞活化更有效,表明靶抗原表位的位置对T细胞活化具有主要影响。Hudecek等也确认,IgG来源的胞外间隔结构域的长度和组成影响CAR-T细胞的功能,而在嵌合抗原设计时,缺少内源性信号功能的胞外间隔结构域对优化体内活性有决定性作用。因此,在将来的研究中,通过控制空间距离增加CAR-T细胞疗法的敏感性是一种很具吸引力的策略。
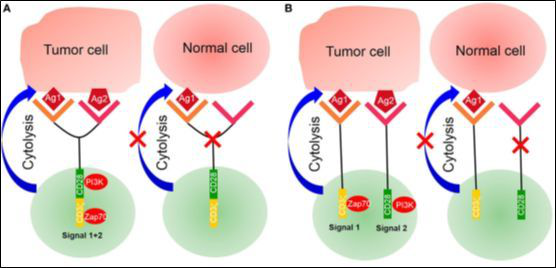
图2 串联嵌合抗原受体(CAR)T细胞
(A)嵌合抗原受体(CAR)的胞外结合结构域由两条不同的肿瘤相关抗原(TAA)特异的单链抗体片段(scFv)组成,并与来源于CD3ζ链和CD28或CD137的胞内信号结构域连接。CAR-T细胞由两个不同的肿瘤相关抗原与各自特异的scFv结合而激活。
(B)嵌合抗原受体的CD3ζ链与共刺激因子一起设计。一个CAR结构包括两个scFv:一个与CD3ζ相连,提供第一信号;另一个与共刺激因子相连,提供第二信号。只有激活了两个信号才能活化T细胞。
之前的研究主要集中在利用外源性活化元件去除MHC分子的限制,而不是内在的T细胞受体。最近,研究人员开发了一种新的CAR-T样细胞,称作双特异T细胞衔接器(BiTE)。这种新设计使用了能分泌T细胞依赖的双特异抗体的转基因T细胞,这种抗体具有两条不同的scFv链,一个识别肿瘤特异的抗原,而另一个识别T细胞特异的抗原(通常是TCR或CD3)。由于这种结构,分泌的scFv可以作为连接肿瘤细胞和T细胞的桥梁,激活BiTE内部的TCR/CD3复合物。但是由于肿瘤细胞上MHC表达不足,尚不清楚T细胞上的CD4或是CD8分子是否参与了此过程。内在的TCR/CD3与分泌的scFv的结合足以传第一信号,而内部的共刺激分子传递第二信号。Luo等开发了能够分泌抗CD3和HER2的双特异抗体,显示出非凡的抗肿瘤效应。有兴趣的是,他们也强调BiTE分泌的双特异抗体影响未转染αHER2/CD3 RNA的“旁观者”T细胞。然而,来自共刺激激动剂的BiTEs的第二活化信号仍不明确。研究者需要使用外源性第二活化信号去增强BiTE的作用。
影响CAR-T细胞疗法的另一个关键因素是T细胞固有的负调控机制。例如,CAR-T细胞成功移动至实体瘤通常上调抑制因子,如程序性死亡因子(Programmed Death-1, PD-1)、细胞毒性T淋巴细胞关联抗原4、T细胞免疫球蛋白与粘蛋白结构域3(T-cell Immunoglobulin Domain and Mucin Domain, TIM3)和淋巴细胞活化基因-3,这些因子特异结合至肿瘤细胞的配基上,从而减缓肿瘤进程。组合免疫治疗策略对改善CAR-T细胞疗法的敏感性是很有前景的。Suarez等将靶向于转移性透明细胞肾细胞癌(ccRCC)上表达的碳酸酐酶IX(CAIX)的CAR-T细胞与程序性死亡配体-1(PD-L1)抗体组合。在其设计中,CAR-T细胞被改造分泌PD-L1抗体,使得局部抗体分布不仅能防止T细胞耗尽,并且能募集NK细胞到肿瘤位点。与单独的抗-CAIX CAR-T细胞相比,人源化小鼠ccRCC模型中的肿瘤生长放慢了5倍,肿瘤重量减少了50-80%。此外,除免疫检查点抑制剂外,越来越多的研究组投身到组合免疫疗法研究中。例如,Junghan等将抗PSMA CAR-T 细胞与IL-2联用治疗前列腺癌,他们发现CAR-T细胞的临床应答受低IL-2浓度血浆的抑制。因此,中等剂量给药的IL-2对增强CAR-T细胞治疗是必需的。该研究也举了一个药物相互作用的药代动力学对二者联用效力的产生关键影响的例子。Curran等建立了通过表达CD40配体(CD40L)增强CAR-T细胞的方法。改造的稳定表达CD40的T细胞(CD40L修饰T细胞)证实在体外细胞增殖和前炎症细胞因子的分泌得到增强。该研究也显示,CD40L修饰的CAR-T细胞诱导树突细胞突变,并分泌前炎症细胞因子IL-12,以增强抗肿瘤作用。因此,在以后的实验设计中,研究人员可以考虑将免疫检查点抑制因子、细胞因子和其他共刺激分子与CAR –T细胞组合。
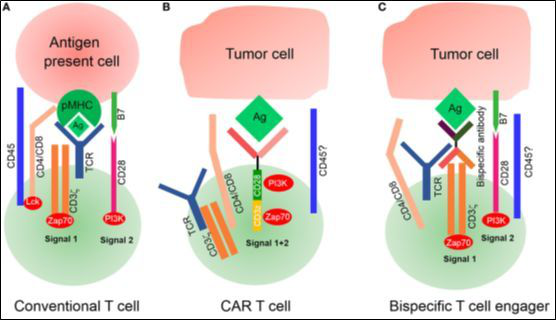
图3 传统T细胞与CAR-T细胞的信号通路
(A)T细胞受体(TCR)与pMHC作用从而形成免疫突触,启动了传统T细胞的活化。T细胞与抗原递呈细胞(APC)的空间距离约为15 nm,因CD45的外功能区过大,物理上将其从免疫突触中排除。 CD4/CD8分子与主要组织相容性复合体(MHC)的I/II募集被CD45磷酸化的淋巴细胞特异性激酶(Lck),激活Zap70提供第一信号。共刺激分子(如CD28)与其在APC上的配体的结合传递第二信号,完成T细胞活化。
(B)改造的CAR-T细胞以非MHC限制的方式通过肿瘤相关抗原(TAA)识别肿瘤细胞。目前尚不清楚CAR-T细胞与肿瘤靶细胞的空间距离,也不清楚空间距离是否小到足以在物理上将磷酸酶CD45从突触中排除。也不清楚嵌合抗原受体(CAR)是否与内源性TCR/CD3或CD4/CD8共同受体作用。
(C)双特异T细胞衔接器(BiTE)能够分泌双特异抗体,其中一个臂可以识别肿瘤相关抗原(TAA)和另一个臂与内在的TCR-CD3复合体相连,但是由于肿瘤细胞上MHC表达不足,尚不清楚CD4/CD8 T细胞是否参与此过程。BiTE通过分泌双特异抗体与靶向表达的细胞连接,内生的TCR/CD3ζ链传递第一信号,而第二信号由BiTE上的内源性共刺激分子与其在肿瘤细胞上的受体传递。BiTE与肿瘤细胞间的空间距离也不受控制,也不清楚CD45是否从免疫突触中排除。
此外,Nishlo和Dottti开发了一种将CAR-T细胞与溶瘤病毒(Oncolytic Viruses, OVs)结合起来的组合疗法,与单独使用CAR-T细胞或溶瘤病毒比较,其抗肿瘤作用更加令人瞩目。他们的研究表明,通过保留对肿瘤细胞的毒性同时不损伤或折损CAR-T细胞活性(即使溶瘤病毒浓度很高时),溶瘤病毒(OV)有益于T细胞的活性。他们证明受溶瘤病毒侵染的肿瘤细胞变得对CAR-T细胞的溶细胞作用更敏感。反过来,肿瘤细胞溶解越快对病毒散播更有利,这强化了实体瘤中的CAR-T细胞。一些研究证明,肿瘤起源的可溶性因子和免疫抑制性免疫细胞在肿瘤微环境中限制了CAR-T细胞的敏感性。这些研究表明在嵌合抗原受体设计时阻断肿瘤微环境中的抑制因子的重要性。在肿瘤微环境中,多种抑制性免疫监测细胞,如髓源性抑制性细胞(Myeloid-derived Suppressor Cells, MDSCs)、调节性T细胞(Treg)、M2和N2表型的肿瘤相关的小噬细胞(Tumor-associated microphages, TAM)或中性粒细胞(Tumor-associated Neutrophils, TAN),构成了抗肿瘤免疫的屏障。MDSc、M2 TAM和N2 TAN是已知的转化生长因子-β(Transform Growth Factor-β, TGF-β)、IL-10、活性氮氧化合物、一氧化氮合酶(Nitric Oxide Synthase, NOS)、精氨酸酶(Arginase, ARG)生成细胞。TGF-β可以诱导多种反应,从组织生长和胚胎形成到上皮细胞的生长抑制和程序性死亡。然而,在肿瘤组织中,越来越多的研究已经确认TGF-β通过抑制CD8细胞毒T淋巴细胞阻断抗肿瘤作用,促进肿瘤细胞迁移和增殖。精氨酸酶和一氧化氮合酶对在肿瘤免疫中具有重要作用的L-精氨酸代谢非常关键。L-精氨酸的出现促进T细胞效应功能和记忆T细胞分化。然而,在一些实体瘤中,多种抑制性免疫监视细胞过表达一种或这两种酶,进而由于精氨酸在肿瘤微环境中的不足,而导致T细胞机能失调。因此,对肿瘤来源的可溶性因子和肿瘤位点中免疫抑制性免疫细胞的活性可能会增强CAR-T细胞治疗的效率。
靶抗原的安全性
嵌合抗原受体T细胞通过识别肿瘤相关抗原(TAA)在肿瘤细胞上的表达攻击其靶细胞。然而,大多数肿瘤相关抗原(TAA)不仅在肿瘤细胞上高度表达,在正常细胞中也有表达。因此,“脱靶效应”引起的毒性风险是实体瘤CAR-T细胞疗法开发的主要障碍。由于肿瘤溶解综合征和“细胞因子风暴”,一些CAR-T细胞疗法引起了生命危险和对胎儿的不良事件。例如,Lamer等评价了用CAIX CAR改造的T细胞治疗转移性肾癌中对靶器官的毒性。在1-2×109总细胞剂量时,12名患者中有4名观察到了一般毒性标准分级2-4级的肝酶紊乱,这种情况可以通过预注射抗CAIX单克隆抗体防止。Morgan等在抗-HER2 CAR的I期临床试验中报道了一种严重的副反应。在第三代(CD28.4-1BB.ζ)抗HER2 CAR临床试验中,向一名具有肺肝原发灶的结肠癌患者注射了1×1010 CAR-T细胞。在15分钟内,该患者出现了急性呼吸窘迫,并于治疗后5天死亡。他们推测低水平表达HER2的肺表皮细胞被注射的细胞识别,启动了“细胞因子风暴”。
通常,安全性与特异性密切相关。在设计嵌合抗原受体(CAR)时,我们需要选择在肿瘤细胞高度表达而在正常细胞中不表达(或低表达)的肿瘤相关抗原(TAA)作为靶标。截止目前,几乎所有实体瘤治疗中的肿瘤相关抗原(TAA)都在正常组织中表达,尤其是周边区域。抗肿瘤作用与CAR-T细胞的剂量有关,高剂量具有毒性增加的潜在风险;因此,很难平衡安全性和有效性。因此,研究人员指出自杀基因系统或许能改善毒性特征。
单纯疱疹病毒胸苷激酶(Herpes Simplex Virus-Thymidine Kinase, HSV-TK)自杀基因系统在细胞和基因治疗中被极普遍地测试,以消除转导细胞潜在的副作用。HSV-TK基因已被成功转染至多种细胞系,使其具有对抗疱疹药物更昔洛韦的敏感性,其有效性在体内和体外均已得到了证明。另一种自杀基因系统是可诱导的半胱天冬酶9(inducible Caspase 9, iCasp9)基因,通常和小分子AP1903联用。iCasp9基因由人Caspase 9蛋白胞内结构域与融合在人FK506结合蛋白药物结合结构域上的凋亡分子组成。这使其在与二聚化药物连接后,可以二聚化并激活凋亡。AP1903的出现使iCasp9蛋白的药物结合结构域产生交联,反过来使Caspase 9二聚化,进而激活下游效应因子Caspase 3,引起细胞凋亡。Diaconu等通过基于Casp9的自杀基因选择性突变设计了一个新的CD19-特异的CAR修饰T细胞(CDa9.CAR Ts)。他们证明iCasp9自杀基因在细胞因子释放综合征中以剂量依赖的方式消耗CD19.CAR-T细胞,或进行正常B细胞重建按要求完全消除。在人源化小鼠模型中,数据也表明低剂量的AP1903能够特异地遏制CD19.CAR-T细胞扩张和细胞因子释放。目前,用于实体瘤的CAR-T细胞受控的特异性安全“开关”控制正在研究之中。
结论
对于人类健康癌症是现实的威胁,CAR-T细胞出现代表抗癌疗法的未来,尤其是对非实体瘤而言。然而,实体瘤的CAR-T细胞疗法面临诸多挑战。应用CAR-T细胞治疗实体瘤的三大障碍是特异性肿瘤相关抗原(TAA)的鉴定、CAR-T细胞难以抵达肿瘤位点,以及肿瘤微环境的免疫抑制效应。在此,我们聚焦于嵌合抗原受体(CAR)的设计,以着手解决这三个问题,增强CAR-T细胞的特异性、敏感性和安全性。
在本文中讨论了克服实体瘤肿瘤微环境的几种方法。研究人员可以将CAR-T细胞疗法与免疫检查点抑制剂组合,或设计靶向免疫检查点的嵌合抗原受体。研究人员也可以设计靶向肿瘤微环境(包括缺氧、营养饥饿、代谢、基质)的嵌合抗原受体。例如,吲哚胺-2,3双加氧酶(Indoleamine 2,3-dioxygenase, IDO)是表达在肿瘤和骨髓细胞上的胞内酶,可以阻断CAR-T细胞的增殖和存活;因此,可以开发靶向IDO的嵌合抗原受体,或肿瘤治疗时组合CAR-T细胞与IDO抑制剂治疗肿瘤。研究人员也鉴定了作为潜在靶点、特异表达在肿瘤细胞上的新抗原。一些研究组也证明趋化因子受体与肿瘤细胞趋化因子配对,能够将CAR-T细胞吸引到肿瘤位点。
CAR-T在恶性血液肿瘤治疗中取得的巨大成功推动了实体瘤CAR-T疗法的发展。对肿瘤发生和肿瘤进程更深入的理解可以促进未来的癌症治疗,并为癌症预防指明希望。
参考文献:
Wang Y, Luo F, Yang J, Zhao C, Chu Y.New Chimeric Antigen Receptor Design for Solid Tumors.Front Immunol. 2017 Dec 22;8:1934.
上一篇:没有了 下一篇:一分钟教你看懂免疫治疗和靶向治疗的差别 |